Différence entre Upgma et le voisin de jointure d'arbre
Le différence clé Entre Upgma et l'arbre de jointure de voisin est le type de l'arbre phylogénétique résultant de chaque méthode. L'UPGMA est la technique de construction d'un arbre phylogénétique enraciné tandis que le voisin se joignant à l'arbre est la technique de construction d'un arbre phylogénétique non racé.
Les arbres phylogénétiques sont des diagrammes en forme d'arbres qui présentent des relations évolutives entre les organismes. Un arbre phylogénétique peut avoir différentes topologies en fonction de la technique utilisée pour la construction d'arbres. L'UPGMA et l'arbre de rejoindre le voisin sont deux méthodes principales qui construisent des arbres phylogénétiques.
CONTENU
1. Aperçu et différence clé
2. Qu'est-ce que Upgma
3. Qu'est-ce que le voisin se joigne à l'arbre
4. Similitudes entre UPGMA et le voisin de jointure d'arbre
5. Comparaison côte à côte - Upgma vs voisine se joignant à l'arbre sous forme tabulaire
6. Résumé
Qu'est-ce que Upgma?
En bioinformatique, il existe différentes techniques de regroupement. Upgma représente Méthode de groupe de paire non pondérée et moyenne arithmétique. C'est une méthode de regroupement hiérarchique. La méthode a été introduite par Sokal et Michener. C'est la technique la plus rapide qui développe un arbre phylogénétique. L'arbre phylogénétique résultant est un arbre phylogénétique enraciné avec un ancêtre commun.
Lors du dessin d'un arbre phylogénétique en utilisant la méthode UPGMA, il considère les taux évolutifs comme le même pour toutes les lignées. Ainsi, c'est une hypothèse importante faite dans la technique UPGMA. Cependant, c'est également le principal inconvénient de la technique car le taux de mutation n'est pas pris en compte pendant la construction d'arbre. Au lieu de cela, il suppose que le taux de mutation est constant. De plus, cette hypothèse est appelée «hypothèse de l'horloge moléculaire». Par conséquent, dans le contexte réel, l'arbre phylogénétique construit à partir d'une méthode UPGMA peut ne pas être précis et fiable.
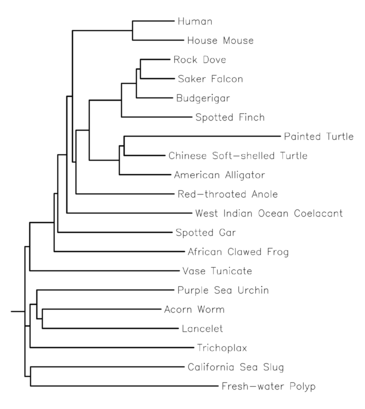
Figure 01: Un arbre phylogénétique tiré de l'UPGMA
La méthode UPGMA considère les distances par paire pour produire un arbre phylogénétique. Initialement, chaque espèce est un cluster, et deux de ces grappes avec la plus petite distance évolutive forment une paire. Par conséquent, cela dépend de la matrice de distance. Les expressions d'algorithme jouent un rôle majeur dans l'interprétation des données d'un arbre phylogénétique dessiné en utilisant la méthode UPGMA.
Qu'est-ce que le voisin se joigne à l'arbre?
Le voisin de jointure d'arbre est une autre technique de regroupement utilisée pour produire un arbre phylogénétique. Naruya Saitou et Masatoshi Nei étaient les pionniers de l'introduction de la méthode. La technique produit un arbre non racé, contrairement à upgma. De plus, le regroupement de cette méthode ne reposait pas sur des distances ultramétriques. Cependant, il considère la variation des taux d'évolution lors de la construction de l'arbre phylogénétique. Ainsi, il existe des variations dans les arbres dessinés en utilisant cette technique. Par conséquent, cette méthode utilise des algorithmes mathématiques spéciaux pour évaluer ces variations.
Figure 02: Un arbre phylogénétique tiré de la méthode de jointure voisine
Lors de la construction des arbres, cette méthode considère les distances entre chaque lignée séparément. Chaque lignée rejoint le nœud nouvellement construit dans l'arbre. Tous ces nœuds rejoignent le nœud central. Par conséquent, lorsqu'un nouveau nœud apparaît, la distance entre le nœud central et le nouveau nœud est importante et est calculé à l'aide des algorithmes. Ces données algorithmiques décident du placement du nouveau nœud.
Quelles sont les similitudes entre UPGMA et le voisin de jointure d'arbre?
- Les deux méthodes utilisent des techniques de clustering lors de la construction d'arbres phylogénétiques.
- De plus, les deux méthodes nécessitent l'utilisation d'algorithmes mathématiques pour interpréter l'arbre phylogénétique.
- Les données de séquence d'ADN jouent un rôle important dans les deux méthodes.
- Les deux méthodes entraînent une méthode de clustering ascendante.
- De plus, l'analyse de grands ensembles de données est possible en utilisant les deux techniques.
- Une analyse statistique des données peut être appliquée pour les deux types d'arbres à l'aide de la méthode de bootstrap.
- Les deux jouent un rôle important dans la classification et l'identification des organismes.
- De plus, les deux méthodes fournissent des données sur les relations évolutives des organismes.
Quelle est la différence entre UPGMA et le voisin de jointure d'arbre?
La principale différence entre UPGMA et l'arbre de rejoindre le voisin repose sur le type d'arbre construit. Ainsi, UPGMA produit un arbre enraciné tandis que un arbre de jointure de voisin produit un arbre non racé. De plus, UPGMA est une méthode moins fiable tandis que l'arbre de rejoindre le voisin est une méthode fiable que UPGMA. Donc, c'est une autre différence entre UPGMA et le voisin de jointure d'arbre.
L'info-graphique ci-dessous résume la différence entre UPGMA et le voisin de jointure d'arbre.
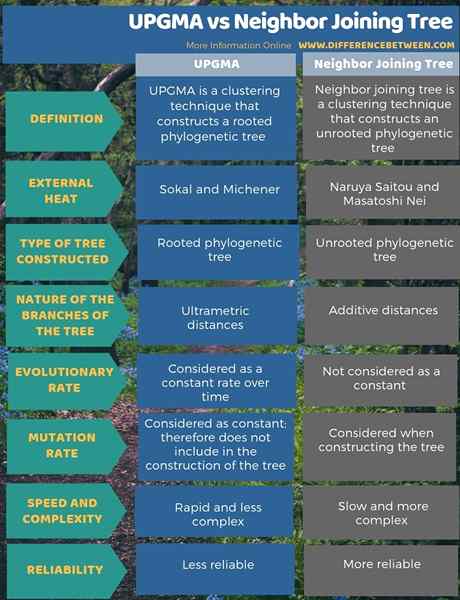
Résumé - Upgma vs voisine rejoignant l'arbre
L'UPGMA et les méthodes des arbres de jointure de voisin sont deux techniques qui sont importantes lors de la construction d'un arbre phylogénétique. Bien que la méthode UPGMA ne tient pas compte du taux d'évolution, la méthode de jointure du voisin le considère pendant la construction d'arbre. Ainsi, la complexité et la fiabilité de l'arbre phylogénétique résultant de la méthode de l'arbre NJ sont élevées. Cependant, ce n'est pas aussi rapide que la méthode UPGMA. De plus, la principale différence entre UPGMA et l'arbre de jointure de voisin s'appuie sur le type d'arbre résultant de chaque technique. L'UPGMA se traduit par un arbre phylogénétique enraciné tandis que le voisin de la méthode de l'arborescence se traduit par un arbre phylogénétique non racé.
Référence:
1. Pavlopoulos, Georgios A, et al. «Un guide de référence pour l'analyse et la visualisation des arbres.»Biodata Mining, Biomed Central, 22 février. 2010, disponible ici.
Image gracieuseté:
1. «CCDC138 Rovered Phylogeny Tree» par KOKXX012 à l'anglais Wikipedia (CC BY-SA 3.0) via Commons Wikimedia
2. «Figure 5» par PLOS One Phylogeny (CC par 2.0) via Flickr