Quelle est la différence entre la phénylcétonurie et la galactosémie
Le différence clé entre la phénylcétonurie et la galactosémie est que la phénylcétonurie est causée en raison de l'accumulation d'un acide aminé appelé phénylalanine dans divers organes du corps, tandis que la galactosémie est causée en raison de l'accumulation de produits chimiques liés au galactose dans divers organes du corps.
La phénylcétonurie et la galactosémie sont deux troubles métaboliques héréditaires. Les troubles métaboliques héréditaires se réfèrent à différents types de conditions médicales causées par des défauts ou des mutations génétiques. Le plus souvent, ils sont hérités des deux parents et interfèrent avec le métabolisme du corps. Ces conditions peuvent également être appelées erreurs innées du métabolisme.
CONTENU
1. Aperçu et différence clé
2. Qu'est-ce que la phénylcétonurie
3. Qu'est-ce que la galactosémie
4. Similitudes - phénylcétonurie et galactosémie
5. Phénylcétonurie vs galactosémie sous forme tabulaire
6. Résumé - Phénylketonurie vs galactosémie
Qu'est-ce que la phénylcétonurie?
La phénylcétonurie est un trouble métabolique rare héréditaire. Il se produit en raison de l'accumulation d'un acide aminé appelé phénylalanine dans divers organes du corps. La phénylcétonurie est causée par un défaut du gène (HAP) qui codes l'enzyme nécessaire pour décomposer la phénylalanine. Sans cette enzyme, une accumulation dangereuse de cet acide aminé se produit lorsque la personne mange des aliments qui contiennent des protéines ou mangent de l'aspartame (un édulcorant officiel). Cela mène finalement à de graves problèmes médicaux. Pour le reste de leur vie, les personnes atteintes de PKU (bébés, enfants et adultes) doivent suivre un régime qui limite la phénylalanine. De plus, les bébés aux États-Unis et de nombreux autres pays sont projetés pour PKU peu de temps après la naissance. Reconnaître la PKU de la bonne façon peut aider à prévenir les problèmes de santé.
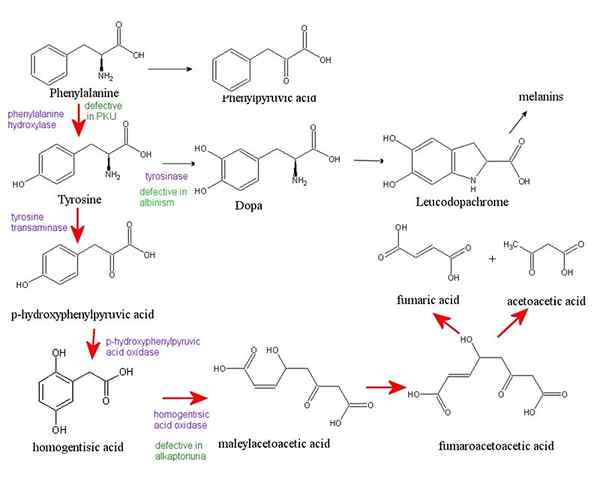
Figure 01: Phénylketonurie
Les signes et symptômes de la PKU peuvent être légers ou graves, et ils comprennent une odeur de moisi dans le souffle, des problèmes neurologiques tels que des crises, des éruptions cutanées, une peau claire et des yeux bleus, une tête anormalement petite, une hyperactivité, une déficience intellectuelle, un développement retardé, un comportement, émotionnel et les problèmes sociaux et les troubles psychiatriques. Cette condition peut être diagnostiquée par antécédents familiaux, tests sanguins et tests génétiques. De plus, un régime à vie avec un apport très limité de protéines, de thérapie à l'acide aminé neutre et de médicaments PKU tels que la saproptérine médicamenteuse (Kuvan) sont les options de traitement pour la PKU.
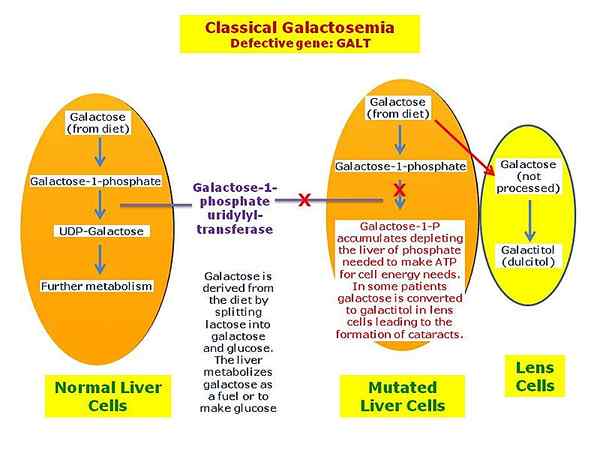
Qu'est-ce que la galactosémie?
La galactosémie est un trouble métabolique rare héréditaire qui se produit en raison de l'accumulation de produits chimiques liés au galactose dans divers organes du corps. C'est une rare erreur innée du métabolisme qui affecte la capacité d'un individu à métaboliser correctement le galactose. La galactosémie suit le mode d'hérédité récessif autosomique, et elle est due à une enzyme responsable d'une dégradation adéquate du galactose. Ce trouble est causé par une carence en enzyme appelée Galactose 1 phosphate uridylyl transférase (GALT) et est due à des mutations dans des gènes tels que Galt, Galk1 et GRAND VENT.
Figure 02: Galactosémie
Les symptômes de la galactosémie peuvent inclure des convulsions, l'irritabilité, la léthargie, une mauvaise alimentation, un mauvais gain de poids, la peau jaune, les yeux blancs (jaunisse) et les vomissements, la diarrhée, les cataractes, les dommages causés par le foie, les problèmes rénaux, les troubles du développement et les ovaires malfaiteurs chez les filles chez les filles. De plus, le diagnostic de cette condition peut être effectué par antécédents familiaux, tests sanguins, tests d'urine, analyse de l'ADN et analyse enzymatique. De plus, l'option de traitement pour la galactosémie comprend un régime faible en galactose. Cela signifie que le lait et les autres aliments contenant du lactose ou du galactose ne peuvent pas être consommés. De plus, l'orthophonie, la planification de l'éducation individuelle et l'intervention, l'hormonothérapie substitutive peut être utile pour traiter d'autres symptômes.
Quelles sont les similitudes entre la phénylcétonurie et la galactosémie?
- La phénylcétonurie et la galactosémie sont deux troubles métaboliques héréditaires ou les erreurs innées du métabolisme.
- Les deux sont dus à des mutations génétiques héréditaires qui provoquent des décences d'enzymes.
- Les deux suivent le mode d'hérédité récessif autosomique.
- Ils sont traitables en restreignant les régimes.
Quelle est la différence entre la phénylcétonurie et la galactosémie?
La phénylcétonurie est un trouble métabolique rare héréditaire causé en raison de l'accumulation d'un acide aminé appelé phénylalanine dans divers organes du corps, tandis que la galactosémie est un trouble métabolique héréditaire rare causé en raison de l'accumulation de produits chimiques liés au galactose dans divers organes du corps. Ainsi, c'est la principale différence entre la phénylcétonurie et la galactosémie. De plus, la phénylcétonurie est due à la mutation du gène HAP, tandis que la galactosémie est due aux mutations de gènes tels que Galt, Galk1, et GRAND VENT.
L'infographie ci-dessous présente les différences entre la phénylcétonurie et la galactosémie sous forme tabulaire pour une comparaison côte à côte.
Résumé - Phénylketonurie vs galactosémie
La phénylcétonurie et la galactosémie sont deux troubles métaboliques héréditaires ou les erreurs innées du métabolisme. La phénylcétonurie se produit en raison de l'accumulation d'un acide aminé appelé phénylalanine dans divers organes du corps, tandis que la galactosémie se produit en raison de l'accumulation de produits chimiques liés au galactose dans divers organes du corps. Ainsi, cela résume la différence entre la phénylcétonurie et la galactosémie.
Référence:
1. «Phénylketonurie.»Nord (Organisation nationale des troubles rares).
2.«Galactosémie: symptômes, causes, diagnostic, traitement.»WebMD.
Image gracieuseté:
1. «Métabolisme de la phénylalanine» par Allen Gathman (CC BY-NC-SA 2.0) via Flickr
2. «Galactosémie» par les troubles (CC BY-SA 4.0) via Commons Wikimedia


