Quelle est la différence entre la SLA et SMA
Le différence clé Entre la SLA et SMA est que la SLA est une maladie des motoneurones qui apparaît la plupart du temps sporadique et est rarement héritée, tandis que SMA est une maladie des motoneurones qui est toujours héréditaire.
La sclérose latérale amyotrophique (SLA) et l'atrophie musculaire spinale (SMA) sont deux types différents de maladies des motoneurones. Ces deux maladies se caractérisent par une perte de motoneurones somatiques et d'innervations aux muscles squelettiques volontaires, entraînant la mort par des échecs de la fonction respiratoire. La SLA est principalement sporadique et peut également être héritée, tandis que SMA est toujours une maladie des motoneurones héréditaires. De plus, la SLA est principalement observée chez les adultes, tandis que la SMA est principalement observée chez les enfants et rarement chez les adultes. De plus, il n'y a pas de remède approprié pour ces deux maladies pour le moment.
CONTENU
1. Aperçu et différence clé
2. Qu'est-ce que la SLA
3. Qu'est-ce que SMA
4. Similitudes - ALS et SMA
5. ALS VS SMA sous forme tabulaire
6. Résumé - ALS vs SMA
Qu'est-ce que la SLA?
La SLA est une maladie neurologique rare qui affecte principalement les nerfs qui sont responsables du contrôle du mouvement musculaire volontaire. Les muscles volontaires sont les muscles que les gens utilisent pour déplacer. La SLA est la forme la plus courante de maladie des motoneurones. Il présente un risque à vie de 1: 300. ALS a normalement l'apparition des adultes et est très agressif. La plupart des patients meurent 2 à trois ans de diagnostic. La SLA semble sporadique dans 90% des cas. Mais il peut être hérité dans 10% des cas.
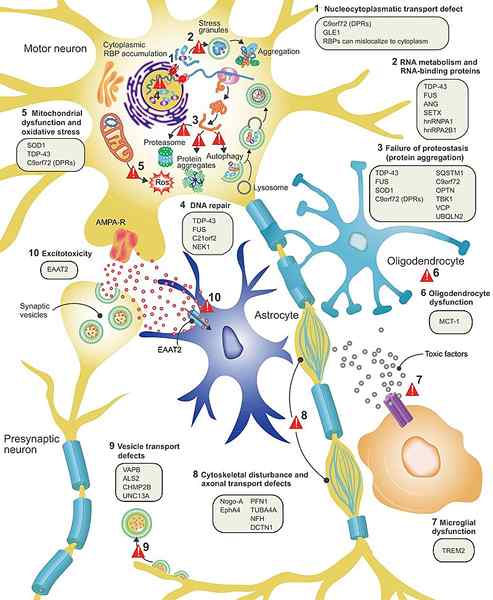
Lorsqu'il est hérité, plusieurs mutations dans différents gènes sont des agents causaux, comme les mutations en superoxyde dismutase 1 (SOD1) gène, Tardbp (TDP43) gène, Fusionné dans le sarcome (Fus) gène et Chromosome 9 Cadre de lecture ouverte 72 (C9ORF72) Gène. Il est également hérité d'un schéma d'hérédité dominante autosomique. Les signes et symptômes de la SLA peuvent inclure des difficultés à marcher, à trébucher et à tomber, à faiblesse souvent dans les jambes, les pieds ou les chevilles, la maladresse, les troubles, les problèmes avalés, les crampes musculaires, les contractions dans les bras, les épaules et la langue, les pleurs, les rires et bâiller de manière inappropriée et des changements dans la fonction cognitive et le comportement.
Figure 01: ALS
La sclérose latérale amyotrophique peut être diagnostiquée par un électromyogramme (EMG), une étude de conduction nerveuse, une IRM, un étude sanguine et urinaire, un robinet vertébral (ponction lombaire) et une biopsie musculaire. En outre, les options de traitement pour la sclérose latérale amyotrophique peuvent inclure des médicaments tels que le riluzole, l'édaravone, le phénylbutyrate de sodium, le taurursodiol et les thérapies telles que la respiration, la physiothérapie, l'ergothérapie, la thérapie vocale, la thérapie nutritionnelle, le soutien psychologique et social.
Qu'est-ce que SMA?
Le SMA est un trouble héréditaire des neurones qui détruit les nerfs du tronc cérébral et de la moelle épinière, qui sont essentiels pour contrôler les activités musculaires squelettiques telles que parler, respirer et avaler. Cela conduit finalement à une faiblesse musculaire et à l'atrophie. SMA montre généralement des modèles d'héritage récessifs autosomiques. Le début de cette maladie se produit avant 6 mois et la létalité à l'âge de 2 ans.
La SMA est également la cause génétique la plus courante de la mortalité infantile, avec une incidence annuelle de 1: 6000 à 1: 10000 naissances vivantes. De plus, l'atrophie musculaire de la colonne vertébrale est causée en raison de mutations dans le Motor de survie 1 (Smn1) Gene, ce qui conduit à de faibles niveaux de protéine SMN. Il existe quatre types de SMA appelés SMA1 (forme sévère), SMA2 (forme intermédiaire), SMA2 (forme légère) et SMA4 (forme adulte). Les symptômes de la SMA peuvent inclure une disquette ou des bras et des jambes faibles, des problèmes de mouvement, des muscles de contraction ou de tremblement, des problèmes dans les os et les articulations, les problèmes de déglutition et les difficultés respiratoires.
L'atrophie musculaire spinale peut être diagnostiquée par des tests sanguins, des tests génétiques, des tests de conduction nerveuse et une biopsie musculaire. En outre, les options de traitement pour l'atrophie musculaire spinale peuvent inclure le traitement modifiant la maladie (donnant aux médicaments stimulant la production de protéines SMN tels que Nusinersen) et la thérapie de remplacement des gènes (perfusion intraveineuse d'un médicament appelé onasemnogene abeparvovec-xioI qui remplace ou défectueux Smn1 gène).
Quelles sont les similitudes entre ALS et SMA?
- ALS et SMA sont deux maladies des motoneurones.
- Les deux maladies affectent principalement le mouvement musculaire volontaire.
- Ce sont des maladies rares.
- Les deux maladies peuvent être héritées.
- Ils peuvent provoquer une létalité sévère.
- Les deux maladies peuvent être identifiées chez les adultes.
- Les deux maladies peuvent avoir des symptômes similaires, tels que des problèmes de mouvement, des problèmes de déglutition et des difficultés respiratoires.
- Ils peuvent être diagnostiqués par examen physique, tests sanguins et conduction nerveuse.
- Ils sont principalement traités par des médicaments spécifiques.
Quelle est la différence entre la SLA et SMA?
La SLA est une maladie des motoneurones qui semble sporadique la plupart du temps et est rarement héritée, tandis que SMA est une maladie des motoneurones qui est toujours héritée. Ainsi, c'est la principale différence entre la SLA et SMA. De plus, la SLA montre un schéma d'hérédité dominant autosomique lorsqu'il est hérédit.
L'infographie ci-dessous présente les différences entre la SLA et le SMA sous forme tabulaire pour une comparaison côte à côte.
Résumé - ALS vs SMA
Les maladies des motoneurones sont des conditions rares qui endommagent progressivement les parties du système nerveux. ALS et SMA sont les deux troubles des neurones les plus courants. La SLA est une maladie des motoneurones qui semble sporadique la plupart du temps et est rarement héritée. C'est autosomique dominant. SMA est une maladie des motoneurones qui est toujours héritée. C'est autosomique récessif. Les deux peuvent entraîner la mort par insuffisance respiratoire. Donc, c'est la principale différence entre la SLA et SMA.
Référence:
1. «Sclérose latérale amyotrophique (SLA).«Mayo Clinic, Mayo Foundation for Medical Education and Research.
2. «Présentation: choix de l'atrophie musculaire de la spinale» NHS Choices, NHS.
Image gracieuseté:
1. «Pathologie de la maladie de la SLA et mécanismes proposés de la maladie» par Philip Van Damme, Wim Robberecht et Ludo van den Bosch - «Modélisation de la sclérose latérale amyotrophique: progrès et possibilités» Modèles et mécanismes de maladie. 10 (5): 537-549. doi: 10.1242 / dmm.029058 PMC: 5451175 PMID: 28468939 (CC par 3.0) via Commons Wikimedia