Quelle est la différence entre le syndrome de Prader Willi et Angelman
Le différence clé Entre Prader Willi et le syndrome d'Angelman est que le syndrome de Prader Willi est causé par la perte de fonction des gènes exprimés paternellement dans une région du chromosome 15 en raison d'une délétion ou d'une désomalie uniparentale tandis à une suppression ou à une désomalie uniparentale.
Les syndromes de Prader Willi et Angelman sont deux troubles génétiques qui peuvent survenir en raison de la perte d'une région chromosomique en raison de la suppression. Ils sont également associés à une désomalie uniparentale, qui fait référence à la situation dans laquelle deux copies d'un chromosome proviennent du même parent au lieu d'une copie provenant de la mère et l'autre copie provenant du père. En outre, ils sont également connus sous le nom de troubles d'impression génétiques (ou génomiques).
CONTENU
1. Aperçu et différence clé
2. Quel est le syndrome de Prader Willi
3. Qu'est-ce que le syndrome d'Angelman
4. Similitudes - Prader Willi et Syndrome d'Angelman
5. Prader Willi vs Angelman Syndrome sous forme tabulaire
6. Résumé - Prader Willi vs Angelman Syndrome
Quel est le syndrome de Prader Willi?
Le syndrome de Prader Willi est un trouble génétique causé par la perte de fonction des gènes exprimés paternellement dans une région du chromosome 15 en raison d'une suppression ou d'une désomalie uniparentale. Dans le syndrome de Prader Willi, environ 74% des cas se produisent lorsque une partie du chromosome 15 du père est supprimée. Dans 25% d'autres cas, la personne affectée a deux copies du chromosome maternel 15 et n'a pas de copie paternelle. Comme certaines parties du chromosome de la mère sont désactivées par une empreinte, elles se retrouvent sans copies de travail de certains gènes.
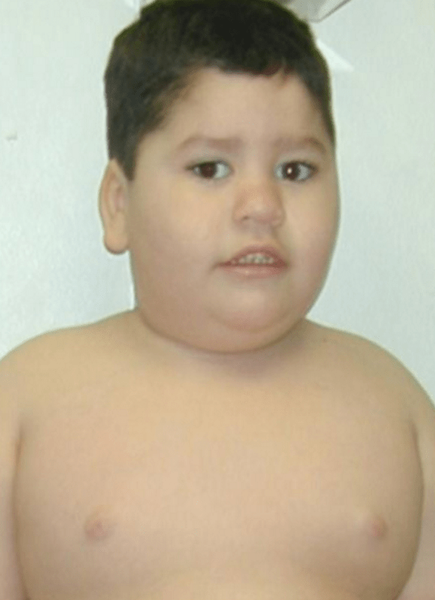
Figure 01: Syndrome de Prader Willi
Les symptômes des nourrissons qui souffrent du syndrome de Prader Willi peuvent inclure un mauvais tonus musculaire, des caractéristiques faciales distinctes, un mauvais réflexe de succion, généralement une mauvaise réactivité et des parties génitales sous-développées. Les signes et symptômes de la petite enfance à l'âge adulte peuvent inclure l'envie de nourriture et la prise de poids, les organes sexuels sous-développés, la mauvaise croissance et le développement physique, les troubles cognitifs, le développement moteur retardé, les problèmes de parole, les problèmes de comportement, les troubles du sommeil, la scoliose, les problèmes de hanche, la salive réduite débit, problèmes de vision, problèmes de température de régulation et manque de pigmentations provoquant des cheveux, des yeux, la peau.
Ce trouble génétique peut être diagnostiqué par des examens physiques, des tests sanguins et des tests génétiques. De plus, les enfants atteints du syndrome de Prader Willi sont traités par une bonne nutrition, un traitement hormonal de croissance humaine, un traitement hormonal sexuel, une gestion du poids, un traitement des troubles du sommeil, diverses thérapies, une thérapie comportementale, des soins de santé mentale et d'autres traitements pour les problèmes de vision, l'hypothyroïdie, le diabète , Scoliose, etc. De nombreux adultes atteints de ce trouble génétique vivent dans des établissements de soins résidentiels, qui leur permettent de manger une alimentation saine, de vivre en toute sécurité, de travailler et de profiter des activités de loisir.
Qu'est-ce que le syndrome d'Angelman?
Le syndrome d'Angelman est un trouble génétique causé par la perte de fonction des gènes exprimés maternellement dans une région du chromosome 15 en raison d'une délétion ou d'une désomalie uniparentale. Le syndrome d'Angelman est dû au manque de fonction d'une région de chromosome 15 causée par une suppression ou une nouvelle mutation (délétion ou mutation de l'UBE3AGENE sur le chromosome 15). Parfois, cela se produit en raison de héritage de deux copies du chromosome 15 du père d'une personne et non de la mère. Comme les copies du père sont inactivées par l'empreinte génomique, aucune version fonctionnelle du gène ne reste.
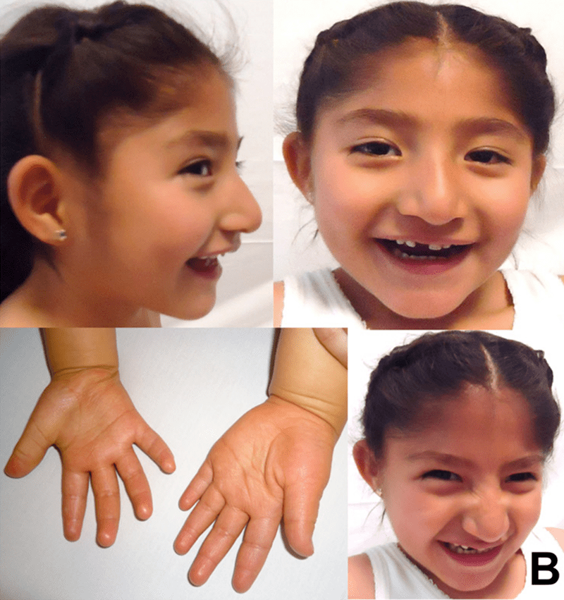
Figure 02: Syndrome d'Angelman
Les signes et symptômes de cette condition peuvent inclure des retards de développement: pas de rampe ou de babillage à 6 à 12 mois, une déficience intellectuelle, pas de discours, des difficultés de marche, un sourire et des rires fréquents, une personnalité heureuse, excitable, une difficulté ou une difficulté d'alimentation, des difficultés à dormir et rester endormi, les crises généralement entre 2 et 3 ans, les mouvements raides ou saccadés, la petite taille de la tête avec une planéité à l'arrière de la tête, la poussée de la langue, les cheveux, la peau et les yeux sont de couleur plus claire, des comportements inhabituels tels que la main battement et bras élevés pendant la marche, les problèmes de sommeil et la colonne vertébrale incurvée.
De plus, ce trouble génétique peut être diagnostiqué par des examens physiques, des tests sanguins, des tests de parole, des tests de motifs d'ADN parental, des tests de chromosomes manquants et des tests de mutation génétique. En outre, les options de traitement pour le syndrome d'Angelman comprennent les médicaments anti-seseure, la physiothérapie, la communication et l'orthophonie, la thérapie comportementale et les médicaments et la formation au sommeil, les changements alimentaires et les médicaments.
Quelles sont les similitudes entre Prader Willi et le syndrome d'Angelman?
- Les syndromes de Prader Willi et Angelman sont deux troubles génétiques qui peuvent être dus à la perte de la région chromosomique due à la suppression ou à la désomalie uniparentale.
- Ils sont également connus sous le nom de troubles d'impression génétique.
- Les deux troubles ne sont pas hérités.
- Ces troubles sont causés à des problèmes dans le chromosome 15.
- Les hommes et les femmes sont également affectés par les deux troubles.
- Ils sont traitables par des soins de soutien.
Quelle est la différence entre le syndrome de Prader Willi et Angelman?
Le syndrome de Prader Willi est un trouble génétique causé par la perte de fonction des gènes exprimés paternellement dans une région du chromosome 15 due à une suppression ou une désomalie uniparentale, tandis que le syndrome d'Angelman est un trouble génétique causé par la perte de fonction des gènes exprimés par maternité dans une région de la région de chromosome 15 en raison d'une suppression ou d'une désomalie uniparentale. Ainsi, c'est la principale différence entre le syndrome de Prader Willi et Angelman. En outre, la prévalence de la maladie pour le syndrome de Prader Willi est une personne sur 15000, tandis que la prévalence de la maladie pour le syndrome d'Angelman est une personne sur 12000 à 20000.
L'infographie ci-dessous présente les différences entre Prader Willi et le syndrome d'Angelman sous forme tabulaire pour une comparaison côte à côte.
Résumé - Prader Willi vs Angelman Syndrome
Les syndromes de Prader Willi et Angelman sont deux troubles génétiques rares. Le syndrome de Prader Willi est un trouble génétique causé par la perte de fonction des gènes exprimés paternellement dans une région du chromosome 15 en raison d'une suppression ou d'une désomalie uniparentale. Pendant ce temps, le syndrome d'Angelman est un trouble génétique causé par la perte de fonction des gènes exprimés par maternité dans une région du chromosome 15 en raison d'une suppression ou d'une désomalie uniparentale. C'est donc la principale différence entre les syndromes de Prader Willi et Angelman.
Référence:
1. «Syndrome de Prader-Willi.«Un aperçu | Sujets ScienceDirect.
2. «Syndrome d'Angelman.«Mayo Clinic, Mayo Foundation for Medical Education and Research, 1 mars. 2022.
Image gracieuseté:
1. «Prader-Willibmc» par Cathetto M, Angulo M, Hertz G, Whitman B. - NCBI (CC par 2.5) Via Commons Wikimedia
2. "Mexican Girl de 5 ans avec le syndrome d'Angelman (Cropped)" par Yokoyama-Rebollar E, Ruiz-Herrera A, Lieberman-Hernández E, Del Castillo-Ruiz V, Sánchez-Sandoval S, Ávila-Flores SM, Castrillo JL - Mol Cytogenet (2015) - Syndrome d'Angelman en raison de la translocation familiale: résultats supplémentaires inattendus caractérisés par une hybridation génomique comparative basée sur des puces à ADN. (CC par 4.0) via Commons Wikimedia