Quelle est la différence entre l'alkaptonurie et la phénylcétonurie
Le différence clé Entre l'alkaptonurie et la phénylketonurie est que l'alkaptonurie est un trouble génétique hérédit.
Les erreurs innées du métabolisme sont des troubles génétiques hérités rares. Dans ces conditions, le corps ne peut pas transformer correctement les aliments en énergie. Les troubles des erreurs innées du métabolisme sont normalement causées en raison de défauts dans des enzymes spécifiques qui aident à décomposer des parties de la nourriture. Il existe de nombreux types d'erreurs innées du métabolisme. Certains d'entre eux sont l'intolérance au fructose, la galactosémie, la maladie d'urine du sucre d'érable, l'alkaptonurie et la phénylcétonurie.
CONTENU
1. Aperçu et différence clé
2. Qu'est-ce que l'Alkaptonurie
3. Qu'est-ce que la phénylcétonurie
4. Similitudes - Alkaptonurie et phénylcétonurie
5. Alkaptonurie vs phénylketonurie sous forme tabulaire
6. Résumé - Alkaptonurie vs phénylketonurie
Qu'est-ce que l'Alkaptonurie?
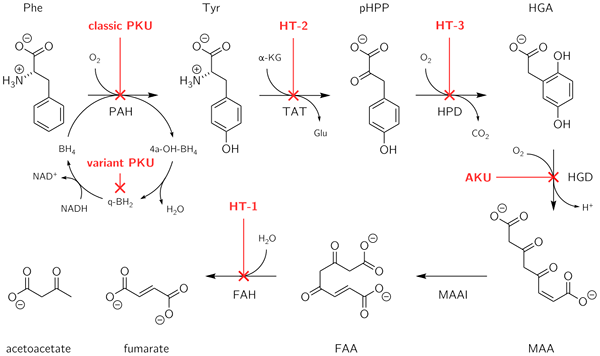
L'alkaptonurie est un trouble génétique héréditaire qui résulte de l'incapacité de métaboliser deux acides aminés: la tyrosine et la phénylalanine. C'est un type d'erreur innée du métabolisme. Ce trouble est dû à une mutation dans un gène appelé HGD, qui est responsable de la création d'une enzyme connue sous le nom de HGD (homogentisate 1, 2-dioxygénase). L'enzyme HGD est impliquée dans le métabolisme des acides aminés aromatiques tyrosine et phénylalanine. Les personnes atteintes de mutation HGD ne peuvent pas métaboliser l'acide homogentisique généré à partir de la tyrosine dans le 4-Maleylacétoacétate. Ce défaut entraîne l'accumulation d'acide homogentisique dans le sang et les tissus. De plus, dans cet état, l'acide homogentisique et sa forme oxydée (alkapton) sont excrétés dans l'urine, ce qui donne une couleur inhabituellement foncée à l'urine.
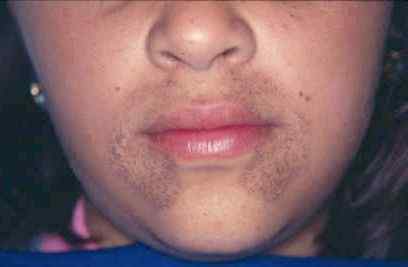
Figure 01: Alkaptonurie
Le signe et les symptômes comprennent les tissus tachés par sombre dans le corps, les problèmes articulaires et osseux (arthrose), l'épaississement et la décoloration bleu-noir du cartilage de l'oreille (ochronose), de la cire d'oreille brun noir ou rougeâtre, des taches brunes ou grises sur les blancs des blancs des blancs les yeux, la sueur décolorée, les zones de peau mouchetées bleues ou noires, les ongles de couleur bleuâtre, les difficultés respiratoires, les problèmes cardiaques (valves cardiaques durcis et les vaisseaux sanguins raides), les calculs rénaux, les pierres de la vessie et les pierres de la prostate. Le diagnostic de cette condition peut être posé par un examen physique, des antécédents détaillés du patient, un test d'urine, une chromatographie en phase gazeuse pour tester les traces d'acide homogentisique et un test d'ADN pour vérifier la mutation du gène HGD. De plus, les options de traitement comprennent le don de la nisiton pour réduire l'acide homogentisique dans le corps, la restriction des protéines dans l'alimentation, l'exercice pour la douleur et la raideur et le soulagement de la douleur par des analgésiques.
Qu'est-ce que la phénylcétonurie?
La phénylcétonurie est un trouble génétique héréditaire qui résulte de l'incapacité de métaboliser un seul acide aminé: la phénylalanine. Cette condition est généralement due à une mutation dans un gène appelé HAP, qui code pour une enzyme connue sous le nom de phénylalanine hydroxylase. Cette enzyme est nécessaire pour métaboliser la phénylalanine d'acide aminé à la tyrosine d'acide aminé. Lorsque l'activité de la phénylalanine hydroxylase se réduit due à la mutation, la phénylalanine s'accumule et est convertie en phénylpyruvate (phénylcélcétone), qui peut être détectée dans l'urine.
Figure 02: Phénylketonurie
Le signe et les symptômes de ce trouble peuvent inclure l'odeur de moisi dans la transpiration, la peau ou l'urine, des problèmes neurologiques, notamment des convulsions, une peau claire et des yeux bleus, une tête anormalement petite, une hyperactivité, une déficience intellectuelle, un développement retardé, un comportement, des problèmes émotionnels, des problèmes sociaux et troubles psychiatriques. Le diagnostic est effectué par des tests sanguins du nouveau-né, une évaluation clinique et un test d'ADN pour la mutation des gènes. En outre, le traitement est principalement par la régulation de l'alimentation, qui comprend des aliments contenant de faibles niveaux de phénylalanine (restriction des protéines) et une formule spéciale pour les bébés avec une petite quantité de lait maternel. Le dihydrochlorhydrate de saproptérine médicament peut également être utile dans certains cas. Ce médicament est un cofacteur pour l'enzyme phénylalanine hydroxylase, ce qui améliore son activité.
Quelles sont les similitudes entre l'alkaptonurie et la phénylcétonurie?
- L'alkaptonurie et la phénylcétonurie sont deux erreurs innées du métabolisme.
- Les deux sont des troubles génétiques hérités.
- Ces troubles suivent l'héritage récessif autosomique.
- Les deux troubles conduisent à l'accumulation de métabolites dans les tissus corporels.
- Ils sont principalement traités en restreignant les régimes riches en protéines.
Quelle est la différence entre l'alkaptonurie et la phénylcétonurie?
L'alkaptonurie est un trouble génétique héréditaire qui résulte de l'incapacité de métaboliser les deux acides aminés, la tyrosine et la phénylalanine, tandis que la phénylcétonurie est un trouble génétique hérédit. Ainsi, c'est la principale différence entre l'alkaptonurie et la phénylcétonurie. En outre, la prévalence mondiale de l'Alkaptonurie est de 1 sur 250000 à 1000000 naissances vivantes, tandis que la prévalence mondiale de la phénylkycétonurie est de 1 sur 23930 naissances vivantes.
L'infographie ci-dessous présente les différences entre l'alkaptonurie et la phénylcétonurie sous forme tabulaire pour une comparaison côte à côte.
Résumé - Alkaptonurie vs phénylketonurie
Les erreurs innées du métabolisme sont des troubles génétiques hérités rares. L'alkaptonurie et la phénylcétonurie sont deux erreurs innées du métabolisme. L'alkaptonurie résulte de l'incapacité de métaboliser deux acides aminés tyrosine et phénylalanine tandis que la phénylcétonurie résulte de l'incapacité de métaboliser l'acide aminé phénylalanine. Ainsi, cela résume la différence entre l'alkaptonurie et la phénylketonurie.
Référence:
1. «Alkaptonurie.»NHS Choices, NHS.
2. «Phénylketonurie (PKU).«Mayo Clinic, Mayo Foundation for Medical Education and Research.
Image gracieuseté:
1. «Ochronose» par Universidad Ces - (CC par 3.0) via Commons Wikimedia
2. «Erreurs innées du métabolisme de la phénylalanine et de la tyrosine» par Bradford Morris - Propre travaux (CC BY-SA 4.0) via Commons Wikimedia