Différence entre l'alpha et la thalassémie bêta
Différence clé - Alpha vs thalassémie bêta
La thalassémie est un groupe hétérogène de troubles causés par des mutations héréditaires qui diminuent la synthèse des chaînes alpha ou bêta globine, conduisant à l'anémie, à l'hypoxie tissulaire et à l'hémolyse des cellules rouges liées au déséquilibre dans la synthèse de la chaîne globine. Il existe deux formes majeures de thalassémie en tant que thalassémie alpha et thalassémie bêta. Dans l'alpha thalassémie, il y a une diminution du nombre de chaînes alpha globin alors que dans la bêta-thalassémie, c'est le nombre de chaînes bêta globines qui diminuent. C'est la principale différence entre l'alpha et la thalassémie bêta.
CONTENU
1. Aperçu et différence clé
2. Quelle thalassémie alpha
3. Quelle thalassémie bêta
4. Similitudes entre l'alpha et la thalassémie bêta
5. Comparaison côte à côte - Alpha vs thalassémie bêta sous forme tabulaire
6. Résumé
Qu'est-ce que la thalassémie alpha?
Dans l'alpha thalassémie, certains des gènes responsables du codage des chaînes alpha globin sont supprimés. Le gène alpha globine en a généralement quatre exemplaires. La gravité de la maladie dépend du nombre de copies manquantes.
Hydrops fœtalis
La synthèse des chaînes alpha globin est complètement supprimée lorsque les quatre copies du gène alpha globine sont manquantes. Étant donné que les chaînes alpha globine sont nécessaires pour la synthèse de l'hémoglobine fœtale et adulte, cette condition n'est pas compatible avec la vie; Par conséquent, l'interruption inutérale de la grossesse se produit si le fœtus est affecté par cette condition.
Maladie HBH
Cette condition est causée par l'absence de trois copies du gène alpha globine. Il en résulte une anémie microcytaire hypochromique modérée à sévère avec une splénomégalie associée.
Traits de thalassémie alpha
Cela est dû à l'absence ou à l'inactivité d'un ou deux copies du gène alpha globine. Bien que les traits alpha thalassémie ne provoquent pas d'anémie, ils peuvent diminuer le volume corpusculaire moyen et les niveaux moyens d'hémoglobine corpusculaire tout en augmentant le nombre de globules rouges sur 5.5 * 1012/ L.
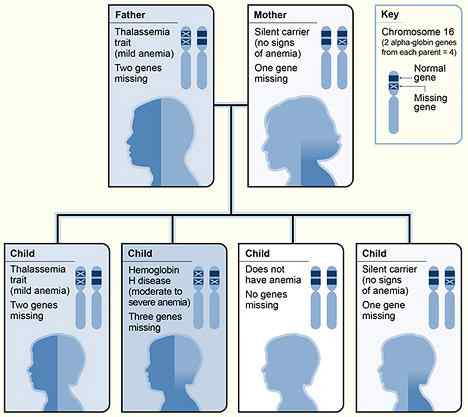
Figure 01: Héritage de la thalassémie alpha
Le diagnostic de la thalassémie alpha se fait par les études de synthèse de la chaîne globine.
Gestion
Les patients présentant une forme légère d'anémie ne nécessitent généralement aucun traitement. L'administration de fer et d'acide folique n'est préconisée que chez certains patients. Ceux qui ont les formes graves de thalassémie alpha ont besoin d'une transfusion sanguine à vie.
Qu'est-ce que la thalassémie bêta?
En thalassémie bêta, la quantité de chaînes bêta globin baisse.
Major à la thalassémie bêta
Si les deux parents sont porteurs du trait de la thalassémie bêta, la possibilité d'une progéniture héritée de la bêta thalassémie majeure est de 25%. Dans la bêta thalassémie majeure, la production de chaînes bêta globine est complètement supprimée, soit considérablement réduite. Puisqu'il n'y a pas assez de chaînes de globines bêta pour qu'elles puissent se combiner, les chaînes en excès alpha globine se déposent dans les globules rouges matures et immatures. Cela conduit à une hémolyse prématurée des globules rouges et à l'érythropoïèse inefficace.
Caractéristiques cliniques
- Anémie sévère, qui devient apparente à 3 à 6 mois après la naissance.
- Splénomégalie et hépatomégalie
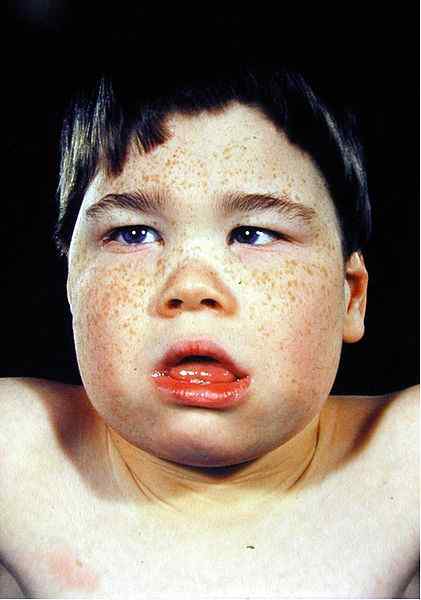
- Faciès thalassémique
Les changements dans les traits du visage sont dus à l'expansion des os en raison de l'hyperplasie de la moelle osseuse. La radiographie à rayons X montre l'apparence de cheveux du crâne qui est généralement observée en bêta thalassémie.
Figure 02: faciès thalassémique
Diagnostic de laboratoire
La chromatographie liquide à haute performance (HPLC) est la principale méthode utilisée dans le diagnostic des maladies hématologiques de nos jours. La thalassémie bêta majeure HPLC montre la présence de niveaux réduits de HBA avec des niveaux inhabituellement élevés de HBF. Une numération sanguine complète révèlera l'existence d'une anémie microcytaire hypochromique, et l'examen d'un film sanguin indiquera la présence d'une quantité accrue de réticulocytes ainsi que des pointillés basophiles et des cellules cibles.
Traitement
- Transfusions sanguines régulières
- Thérapie de chélation en fer
- Acide folique (si l'apport alimentaire de l'acide folique n'est pas satisfaisant)
- Splénectomie (parfois utilisée pour réduire les besoins sanguins)
- Une greffe de moelle osseuse
- Thérapie génique à des fins de dépistage et thérapeutiques
Trait de thalassémie bêta / mineur
La thalassémie bêta mineure est une condition courante qui est souvent sans symptôme. Bien que les signes et les symptômes soient similaires à ceux de la thalassémie alpha, la bêta thalassémie est plus grave que son homologue. Le diagnostic de la bêta thalassémie mineure est posé si le HBA2 Le niveau est supérieur à 3.5%.
Thalassémie intermédia
Thalassemia intermedia fait référence aux cas de thalassémie de gravité modérée qui n'ont pas besoin de transfusions régulières.
Quelle est la similitude entre l'alpha et la thalassémie bêta?
- Dans les deux conditions, il y a une diminution du taux de sang de l'hémoglobine.
Quelle est la différence entre l'alpha et la thalassémie bêta?
Alpha vs thalassémie bêta | |
| Il y a une diminution du nombre de chaînes alpha globin. | Il y a une diminution du nombre de chaînes de bêta globine. |
| Suppression des gènes | |
| Certains des gènes responsables du codage des chaînes alpha globin sont supprimés. | Les gènes qui sont responsables de la synthèse des chaînes bêta globin sont soit partiellement ou complètement supprimés. |
| Les types | |
| Hydrops fetalis, la maladie HBH et les traits de thalassémie alpha sont les principales formes de thalassémie alpha. | Il existe deux principales formes de thalassémie bêta en tant que bêta thalassémie majeure et thalassémie bêta mineure. |
| Diagnostic | |
| Le diagnostic de la thalassémie alpha se fait par les études de synthèse de la chaîne globine. | La chromatographie liquide à haute performance (HPLC) est l'enquête utilisée pour le diagnostic de la bêta thalassémie. |
| Caractéristiques cliniques | |
|
|
| Traitement et gestion | |
|
|
Résumé - Alpha vs Beta Thalassemia
La thalassémie est un groupe hétérogène de troubles causés par des mutations héréditaires qui diminuent la synthèse des chaînes alpha ou bêta globine qui composent l'hémoglobine HBA adulte. La thalassémie peut être classée largement en deux catégories principales en tant que thalassémie alpha et thalassémie bêta. Dans la thalassémie alpha, la quantité de chaînes alpha est diminuée et en bêta-thalassémie, le nombre de chaînes bêta est diminuée. C'est la principale différence entre l'alpha et la thalassémie bêta.
Télécharger la version PDF de la thalassémie alpha vs bêta
Vous pouvez télécharger la version PDF de cet article et l'utiliser à des fins hors ligne selon la note de citation. Veuillez télécharger la version PDF ici différence entre Alpha et Beta Thalassemia
Référence:
1.Kumar, Parveen J., et Michael L. Clark. Médecine clinique de Kumar & Clark. Édimbourg: W.B. Saunders, 2009.
Image gracieuseté:
1. «Thalassemia alpha» par National Heart Lung and Blood Institute (NIH) - National Heart Lung and Blood Institute (NIH) (Domain public) via Commons Wikimedia
2. «ATR-X» par Gibbons R. - Gibbons R. Alpha Thalassaemia. Orphembre j rare dis. 1, 15. 2006. doi: 10.1186 / 1750-1172-1-15. PMID 16722615 (CC par 2.0) via Commons Wikimedia